Introduction
DNA methylation is a pivotal epigenetic regulation mechanism facilitated by DNA methyltransferases (DNMTs). This process involves the covalent bonding of a methyl group at the carbon-5 position of cytosine, resulting in the formation of 5-methylcytosine (5mC). Alongside its derivatives like the “sixth base” 5-hydroxymethylcytosine and 5-formylcytosine, DNA methylation dynamically influences gene expression and demethylation through TET dioxygenases. This regulation plays a crucial role in embryonic development, tumor formation, aging, and is closely linked to conditions such as cancer and neurodegenerative diseases.
Over the past three decades, DNA methylation detection technologies have undergone significant transformation. Initially, these methods relied on chromatography to differentiate between methylated and unmethylated cytosine. Subsequent techniques involved indirect detection through methylation-sensitive restriction enzymes and 5mC antibody immunoprecipitation. A breakthrough in 1992 with bisulfite conversion technology enabled the chemical transformation to distinguish methylation sites, facilitating the first single-base resolution analysis.
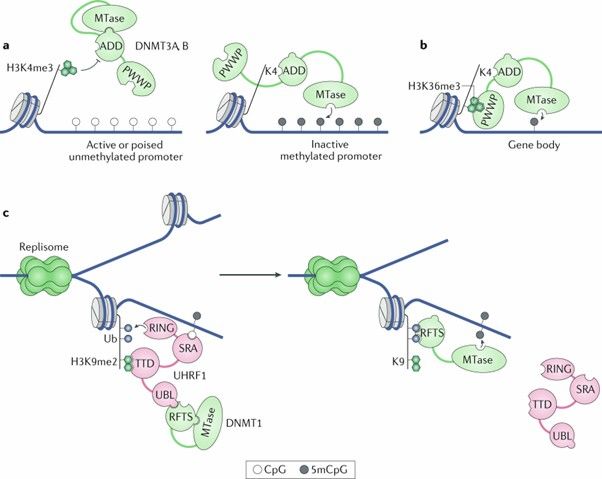
The diverse roles of DNA methylation in mammalian development and disease. (Greenberg, M.V.C., et al., 2019)
More recently, high-throughput technologies such as methylation microarrays (like the Infinium 850K) and third-generation sequencing (including nanopore sequencing) have accelerated the mapping of whole genome epigenomes. Meanwhile, targeted detection techniques such as pyrosequencing and digital PCR show potential for early cancer screening clinical applications.
Yet, significant differences exist among these methods. Whole-genome bisulfite sequencing (WGBS), though considered the “gold standard,” faces challenges like DNA degradation and high costs. Methylation microarrays offer broad compatibility, supporting FFPE samples and are preferred for large cohort studies. Emerging enzyme-based methods (EM-seq) and single-cell sequencing techniques (scWGBS) address issues related to small sample quantities and cellular heterogeneity.
This article systematically explores the biological significance of DNA methylation and its detection methodologies, covering their technical principles, application scenarios, and research strategies. It aims to provide a methodological reference for basic research, focusing on the necessity, technical comparisons, experimental design, and case studies to aid readers in selecting suitable research tools.
Functionality of DNA Methylation Modifications
DNA methylation operates through three primary regulatory models:
1. Classical Model of DNA Methylation Regulation: In this traditional framework, methylated CpG island promoters attract transcription-repressive MBD proteins, preventing transcription factor binding. Conversely, unmethylated CpG islands are accessible to transcription factors.
2. New Model of DNA Methylation for Transcription Regulation: Genes with methylated CpG island promoters are inhibited by repressive complexes containing MBDs. Additionally, enhancer methylation can block transcription factor attachment. Active genes featuring unmethylated CpG island promoters often associate with activator complexes that include a CXXC domain. Transcription factors may also bind to non-methylated enhancers. Furthermore, highly methylated gene bodies of active genes aid in suppressing inadvertent transcription.
3. Decoupling of DNA Methylation from Transcription Initiation Inhibition: In certain scenarios, promoters with low-density CpG methylation undergo active transcription. Repressive MBD proteins do not engage with these promoters, though the underlying reasons remain unclear. Additionally, sequences with low-density CpG, including enhancers and promoters, may facilitate binding by activating transcription factors. The H3K4me3 histone mark is an example associated with active transcription processes.
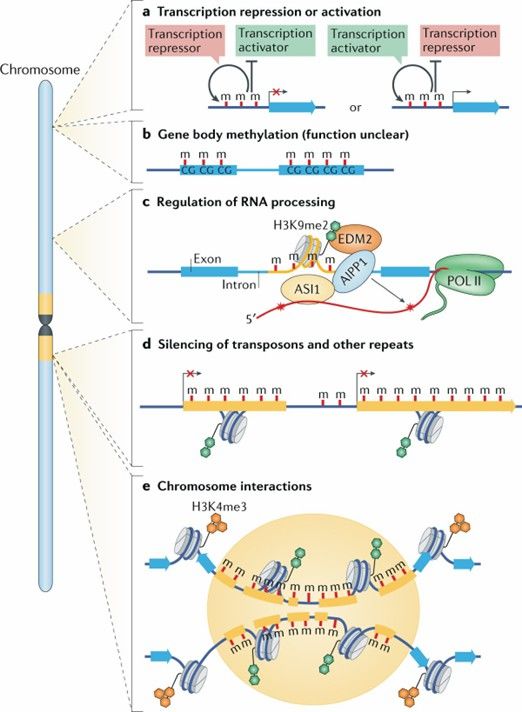
Dynamics and function of DNA methylation in plants. (Zhang, H., et al., 2018)
How much DNA methylation occurs
To effectively analyze human DNA methylation, it’s essential to understand the fundamental aspects of methylation within the human genome. Humans possess approximately 3 billion base pairs, and DNA methylation predominantly occurs at CpG dinucleotides. The genome contains roughly 28 million CpG sites, with 60-80% of these sites typically methylated. CpG islands, which are CpG-rich sequences often found in promoter regions, are usually unmethylated. However, in tumor cells, the overall level of methylation decreases to 20-50% compared to normal cells. This change implies that the methylation status of about 2.8 to 16.8 million CpG sites is altered. In essence, while global methylation levels drop in tumor cells, there’s a notable increase in methylation levels at the CpG sites within gene promoter regions.
Recommended Reading on DNA Methylation
For foundational insights into human and mouse basic medical research and cellular biology, consider the following reviews:
- “DNA methylation in mammalian development and disease.” Nat Rev Genet. 2024
- “DNA methylation: old dog, new tricks?” Nat Struct Mol Biol. 2014, 21(11): 949
For insights into plant biology:
- “Epigenetic gene regulation in plants and its potential applications in crop improvement.” Nat Rev Mol Cell Biol. 2024
- “Non-canonical RNA-directed DNA methylation.” Nat Plants. 2016, 2(11): 16163
Why Detect DNA Methylation
Before we delve into the reasons for detecting DNA methylation, it’s important to ask: what questions can DNA methylation profiling answer? DNA methylation is well-known for its ability to regulate gene expression. Thus, analyzing DNA methylation can provide valuable insights into the regulatory mechanisms governing gene expression. DNA methylation plays a role in a variety of crucial biological processes, including early embryonic development, genomic imprinting, X-chromosome inactivation, silencing of repetitive sequences, and the development and metastasis of cancer. Furthermore, DNA methylation serves as a significant biomarker for tumors.
1. Uncovering the “Hidden Rules” of Gene Expression
DNA methylation finely tunes cell functions by repressing or activating gene transcription:
- Gene Silencing: High methylation levels in promoter regions (e.g., in tumor suppressor genes) prevent transcription factor binding, leading to gene silencing.
- Gene Activation: Low methylation in gene bodies may enhance transcription elongation, supporting cell-specific functions, such as the expression of synaptic proteins in neurons. This “on-off” mechanism explains why cells with identical DNA sequences, like skin and liver cells, perform vastly different tasks.
2. Key Clues to Life’s Mysteries
DNA methylation is integral to numerous core biological processes:
- Embryonic Development: After fertilization, genomes undergo widespread methylation erasure and reconstruction to ensure stem cell pluripotency. For example, abnormal methylation of the imprinted gene H19 can lead to developmental abnormalities in the fetus.
- Genomic Imprinting: Selective expression of maternal or paternal genes (like in Prader-Willi syndrome) relies on methylation marks.
- X-Chromosome Inactivation: In female cells, the silencing of one X chromosome is driven by the methylation of the XIST gene promoter.
- Cancer Metastasis: Tumor cells acquire invasive capabilities through methylation reprogramming of metastasis-related genes, such as TWIST1.
3. The “Molecular Probe” for Cancer Early Screening and Diagnosis
DNA methylation markers are ideal targets for tumor detection due to their high stability and tissue specificity:
- Non-invasive Screening: The detection of Septin9 gene methylation in the blood of colorectal cancer patients is clinically applied, offering early warning without the need for colonoscopy.
- Precise Typing: The methylation level of CDO1 in lung cancer can differentiate between adenocarcinoma and squamous cell carcinoma, guiding treatment decisions.
- Efficacy Monitoring: In breast cancer patients, dynamic changes in the methylation of circulating tumor DNA (ctDNA) post-treatment can provide real-time insights into drug resistance.
Summary of DNA Methylation Detection Methods
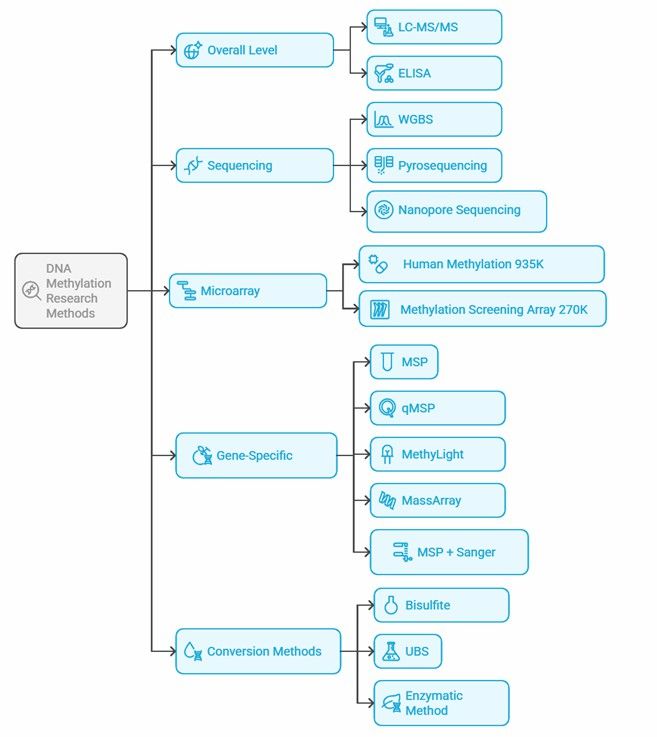
Mind map of DNA methylation research methods.
DNA methylation detection methods vary widely, and the choice depends on the specific research objectives, sample types, and cost considerations. Here’s a concise overview of the primary methods:
1. Global Level Detection:
- LC-MS/MS: Highly accurate and sensitive, considered the “gold standard,” but costly and complex to operate.
- ELISA: Fast and cost-effective but prone to interference and lower sensitivity.
2. Sequencing Technologies:
- WGBS: Provides single-base resolution, ideal for comprehensive studies, though complex and expensive.
- Pyrosequencing: Quantifies methylation in short fragments, suitable for quantitative analysis but with operational complexities.
- Nanopore Sequencing: Directly detects methylation without chemical conversion, promising but still developing and costly.
3. Microarray Technology:
- Used for large-scale, targeted analysis, more affordable than WGBS, and commonly used in clinical and cohort studies.
4. Specific Gene Detection:
- MSP: Simple and inexpensive, ideal for specific gene sites but involves complex primer design.
- MethyLight: Offers high-precision quantification at specific sites using probe-based methods.
- Digital PCR: More sensitive than MethyLight, effective for low methylation levels.
- HRM Analysis: Fast detection of methylation differences, requires precise temperature control.
- PCR + Sanger Sequencing: Confirms methylation sites with high accuracy, the “gold standard.”
- MassArray (MALDI-TOF): High throughput, cost-effective, differentiates between methylated and unmethylated fragments after bisulfite conversion.
Detailed Overview of DNA Methylation Detection Methods
This section provides an overview of various DNA methylation detection methods, including techniques for assessing global methylation levels, sequencing-based approaches, microarray technologies, and gene-specific detection methods.
Global Methylation Level Detection
To assess methylation levels across the entire genome, the following methods can be employed:
1. Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS):
- This method digests genomic DNA into single nucleotides (such as A, T, G, C, 5mC, and 5hmC) and uses mass spectrometry in Multiple Reaction Monitoring (MRM) mode to measure each base’s content. This allows calculation of DNA methylation levels (5mC%) and simultaneous detection of 5hmC. Despite its extremely high accuracy and sensitivity, making it the gold standard for detecting whole-genome 5mC content, LC-MS/MS is expensive and complex to operate, thus unsuitable for high-throughput studies.
2. ELISA Methylation Detection:
- This method utilizes antibodies to detect 5mC levels and, via a standard curve, calculate DNA methylation levels (5mC%). ELISA is simple to use, fast, and available as commercial kits, but its reliance on antibody specificity may limit sensitivity and accuracy, especially in samples with subtle methylation changes.
Sequencing-Based Methylation Detection
To obtain site-specific and sequence-level information on DNA methylation, sequencing methods are ideal, some DNA methylation sequencing methods are described below:
1. Whole Genome Bisulfite Sequencing:
- WGBS combines bisulfite conversion with high-throughput sequencing to detect DNA methylation at single-base resolution across the entire genome. It covers all methylation sites and can be coupled with targeted techniques to detect low-frequency methylation signals, avoiding issues with repetitive sequences and SNPs. As sequencing costs decline and technology advances, WGBS will become more prevalent.
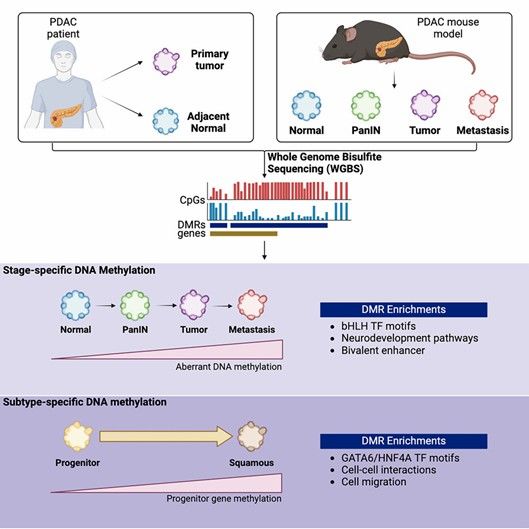
Whole-genome bisulfite sequencing identifies stage- and subtype-specific DNA methylation signatures in pancreatic cancer. (Wang, Sarah S., et al., 2024)
2. Oxidative Bisulfite Sequencing (oxBS-seq):
- By combining traditional BS-seq with chemical oxidation using potassium ruthenate (KRuO₄), this method oxidizes 5hmC to 5fC, which, along with unmodified C, converts to U upon bisulfite treatment, while 5mC remains unchanged. The combined use of oxBS-seq and BS-seq can also achieve single-base resolution detection of 5hmC.
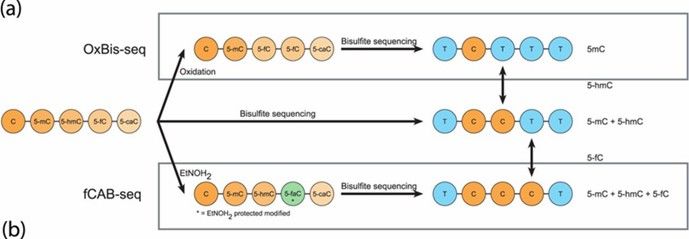
Principal scheme of oxBS-seq and fCAB-seq methods. (Becker, Daniel, et al., 2014)
3. Reduced Representation Bisulfite Sequencing (RRBS):
- RRBS employs restriction enzymes (like MspI) to selectively cut the genome, enriching promoter regions and CpG islands for bisulfite sequencing. This approach increases sequencing depth in high-CpG regions, reducing costs and making it suitable for large-scale clinical research.
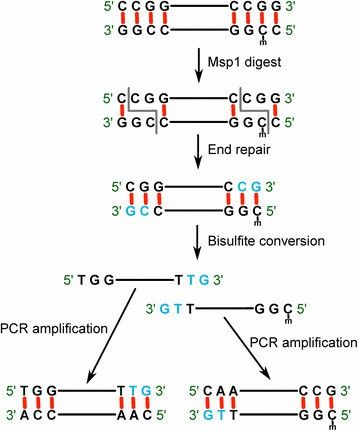
RRBS concept and workflow. (Baheti, Saurabh, et al., 2016)
4. Single-Cell Whole Genome Methylation Sequencing (scWGBS):
- With challenges in library construction for single-cell DNA methylation, new scWGBS technology utilizes linear amplification and single-tube library construction to reduce bias, enabling high-precision methylation analysis for rare samples.
5. Amplicon Methylation Sequencing:
- Designed for specific gene regions using methylation-specific primers, this method employs PCR amplification of bisulfite or oxidized treated DNA, followed by high-throughput sequencing to analyze methylation status, suitable for precise target gene methylation analysis.
6. Hydroxymethylated DNA Immunoprecipitation Sequencing (hMeDIP-seq):
- Using antibodies specific to 5hmC, this method enriches hydroxymethylated DNA fragments for high-throughput sequencing, suitable for studying the role of 5hmC in gene regulation, disease, and embryonic development.
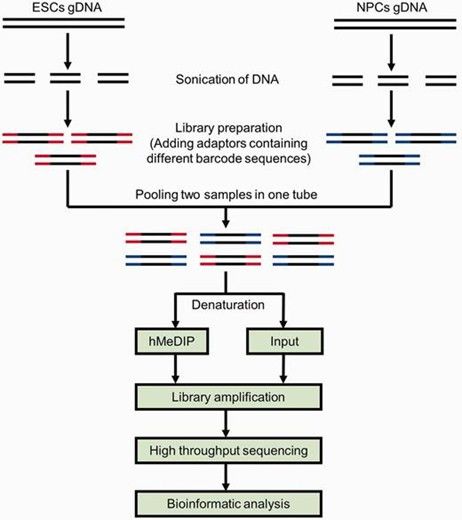
Schematic diagram of the comparative hMeDIP-seq method. (Tan, Li, et al., 2013)
7. Enzymatic Methylation Sequencing (EM-seq):
- EM-seq overcomes WGBS limitations of DNA degradation from bisulfite by using TET2 enzymes and oxidation enhancers to convert 5mC and 5hmC to 5caC, then processing with APOBEC deaminase for methylation detection. This protects DNA integrity and enhances data quality.
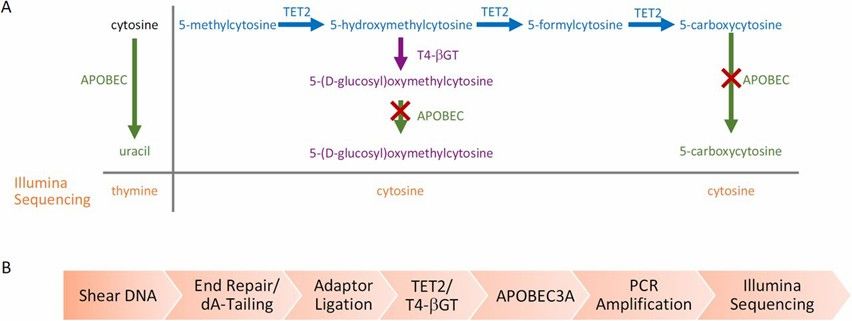
Enzymatic Methyl-seq mechanism of action and workflow. (Vaisvila, Romualdas, et al., 2021)
8. Pyrosequencing:
- Combining bisulfite treatment with pyrosequencing, this method detects methylation in specific target regions by comparing C and T ratios at individual sites. Fast and straightforward, pyrosequencing is ideal for short fragment analysis (20-50bp) but struggles with long homopolymer regions.
9. TET-Assisted Pyridine Borane Sequencing (TAPS):
- TAPS eschews bisulfite treatment by using TET1 oxidase to oxidize 5mC and 5hmC into 5caC, which is then reduced by pyridine borane to DHU, ultimately converting C to T during PCR. TAPS is non-destructive, reducing DNA loss and improving data quality at lower costs compared to traditional WGBS.
10. Nanopore Sequencing:
- This technique directly distinguishes methylated from unmethylated bases via changes in electric signals as DNA passes through nanopores, avoiding bisulfite treatment. Offering simultaneous methylation and sequence data, nanopore sequencing supports long-read sequencing. However, it remains costly, with ongoing improvements needed in data analysis.
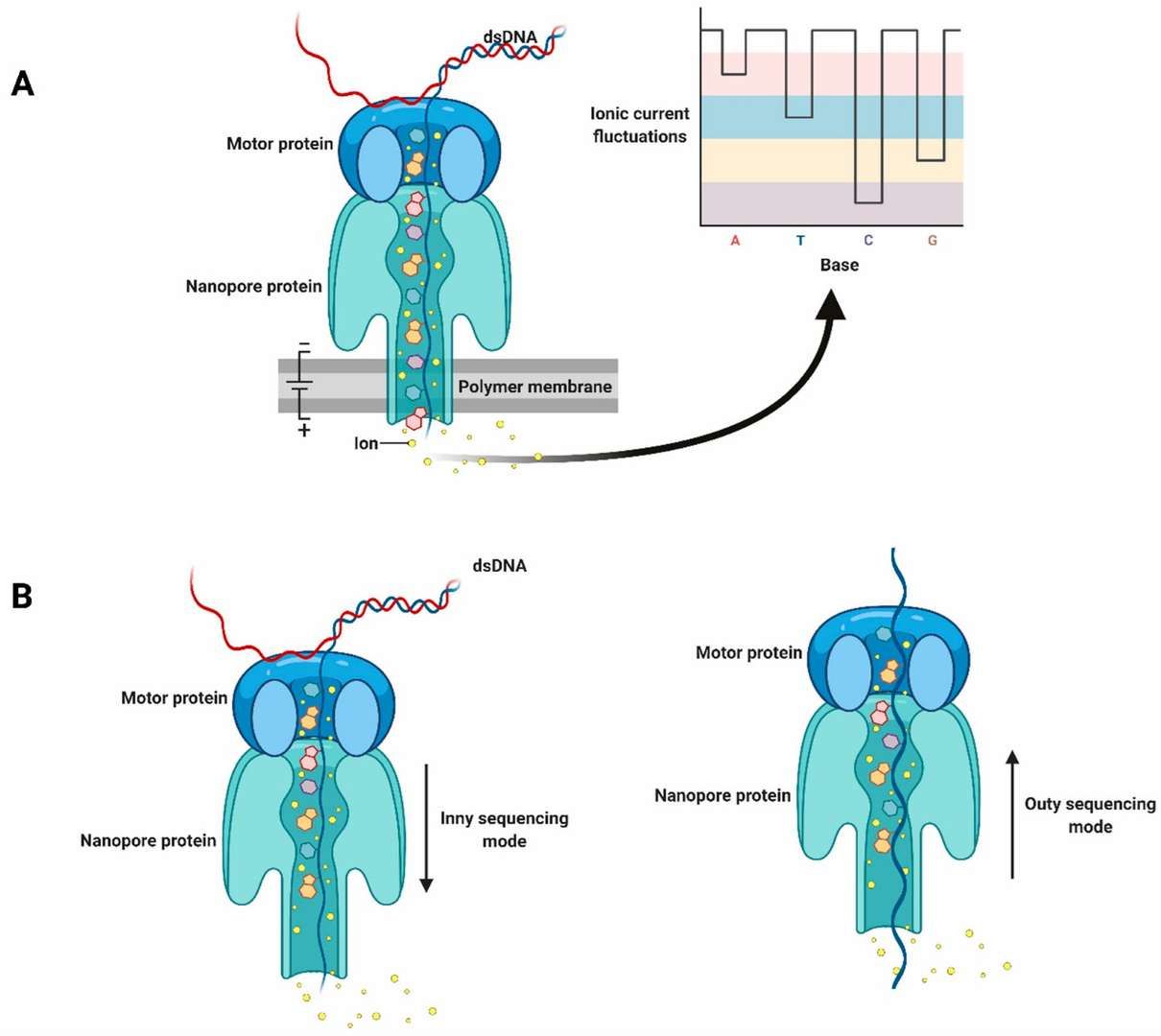
Schematic diagram of nanopore sequencing technology. (Javaran, Vahid Jalali, et al., 2021)
DNA Methylation Microarrays
In large-scale cohort studies of whole-genome DNA methylation, the most feasible approach is often microarray technology rather than WGBS, primarily due to the high costs associated with sequencing and data processing. DNA methylation microarrays leverage hybridization of bisulfite-treated DNA probes, targeting CpG-rich cytosines of interest (both methylated and unmethylated).
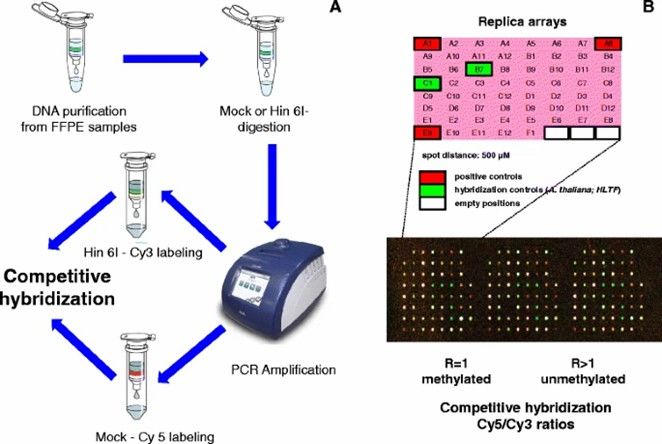
Microarray-mediated methylation assay. (Li, Y., Melnikov, A.A., et al., 2015)
The 450K array, for example, involves the bisulfite conversion of 0.5-1μg of genomic DNA. Converted DNA hybridizes with an array of pre-designed methylation-specific probes: one set targets methylated cytosines, and another targets unmethylated cytosines. The probe’s single base at the 3′ CpG site incorporates a labeled and fluorescently tagged nucleotide (ddNTP). An Illumina iScan scanner then scans the bead array, detecting the fluorescence signal ratio.
The DNA methylation proportion at each CpG site is quantified as a β value, calculated using the following formula:
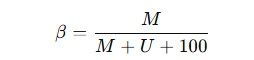
Where ( M ) is the intensity of the methylated signal, ( U ) is the intensity of the unmethylated signal, and 100 is a constant offset for adjustment when signal intensities are low. The β value ranges from 0 (0% methylation) to 1 (100% methylation).
DNA methylation microarrays are vital tools for studying DNA methylation. Compared to whole-genome methylation sequencing, methylation microarrays offer advantages such as lower starting material requirements, shorter processing times, and flexibility in sample types. They can be applied to FFPE (formalin-fixed paraffin-embedded) samples and valuable clinical specimens, thus playing a significant role in methylation detection technology.
To date, the Infinium Human Methylation 450K and Methylation EPIC v1.0 (850K) arrays have been widely adopted by researchers to gather epigenomic data, with increasing numbers of related publications each year. In 2023, Illumina launched the 935K array, covering over 935,000 marker sites. The 935K array retains compatibility with the 850K while removing 107,000 non-functional probes caused by SNPs, cross-hybridization, and multi-mapping, and adding at least 186,000 new probes, providing a more robust tool for clinical DNA methylation research.
Additionally, the Infinium Methylation Screening Array 270K is another DNA methylation chip from Illumina, covering approximately 270,000 CpG sites. Designed for rapid screening, it enables the analysis of DNA methylation in large clinical sample sets, suitable for preliminary methylation screening and validation. Although it has lower resolution, it remains effective for initial studies and high-throughput analysis.
It’s important to note that DNA methylation microarrays are predominantly used for human samples. When dealing with large-scale clinical samples and well-defined research objectives, DNA methylation microarray technology is the most suitable method.